
Reduction potential predictions of some aromatic nitrogen-containing molecules†
Abstract
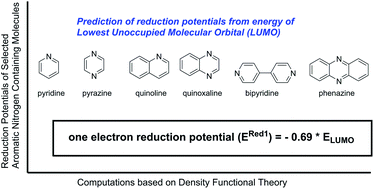
Accurate quantum chemical methods offer a reliable alternative to time-consuming experimental evaluations for obtaining a priori electrochemical knowledge of a large number of redox active molecules. In this contribution, quantum chemical calculations are performed to investigate the redox behavior of quinoxalines, a promising family of active materials for non-aqueous flow batteries, as a function of substituent group. The reduction potentials of 40 quinoxaline derivatives with a range of electron-donating and electron-withdrawing groups are computed. Calculations indicate the addition of electron-donating groups, particularly alkyl groups, can significantly lower the reduction potential albeit with a concomitant decrease in oxidative stability. A simple descriptor is derived for computing reduction potentials of quinoxaline derivatives from the LUMO energies of the neutral molecules without time-consuming free energy calculations. The relationship was validated for a broader set of aromatic nitrogen-containing molecules including pyrazine, phenazine, bipyridine, pyridine, pyrimidine, pyridazine, and quinoline, suggesting that it is a good starting point for large high-throughput computations to screen reduction windows of novel molecules.
 Please wait while we load your content...
Please wait while we load your content...

