
Electronic structure and band alignment of zinc nitride, Zn3N2
Abstract
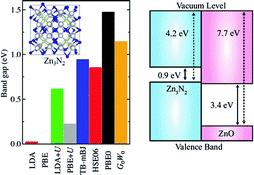
Zinc nitride (Zn3N2) is a promising candidate for optoelectronics applications due to its high electron mobility and high electrical conductivity. It is also thought that Zn3N2 can be used as a starting material to achieve p-type conductivity in ZnO-based oxide homojunctions. In this work, the electronic structure of bulk Zn3N2 is studied using density-functional theory (DFT) with different approximations to the exchange-correlation functional, ranging from (semi-)local functionals to the quasiparticle G0W0 approach. We predict a bandgap in the range of 0.9–1.2 eV, reconciling the scattered values reported in experiments, and a remarkably low work function (ionisation potential) of 4.2 eV for the (111) surface.
 Please wait while we load your content...
Please wait while we load your content...

