
Controlling photoinduced electron transfer from PbS@CdS core@shell quantum dots to metal oxide nanostructured thin films†
Abstract
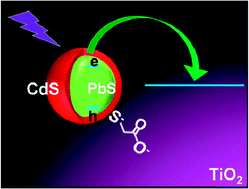
N-type metal oxide solar cells sensitized by infrared absorbing PbS quantum dots (QDs) represent a promising alternative to traditional photovoltaic devices. However, colloidal PbS QDs capped with pure organic ligand shells suffer from surface oxidation that affects the long term stability of the cells. Application of a passivating CdS shell guarantees the increased long term stability of PbS QDs, but can negatively affect photoinduced charge transfer from the QD to the oxide and the resulting photoconversion efficiency (PCE). For this reason, the characterization of electron injection rates in these systems is very important, yet has never been reported. Here we investigate the photoelectron transfer rate from PbS@CdS core@shell QDs to wide bandgap semiconducting mesoporous films using photoluminescence (PL) lifetime spectroscopy. The different electron affinity of the oxides (SiO2, TiO2 and SnO2), the core size and the shell thickness allow us to fine tune the electron injection rate by determining the width and height of the energy barrier for tunneling from the core to the oxide. Theoretical modeling using the semi-classical approximation provides an estimate for the escape time of an electron from the QD 1S state, in good agreement with experiments. The results demonstrate the possibility of obtaining fast charge injection in near infrared (NIR) QDs stabilized by an external shell (injection rates in the range of 110–250 ns for TiO2 films and in the range of 100–170 ns for SnO2 films for PbS cores with diameters in the 3–4.2 nm range and shell thickness around 0.3 nm), with the aim of providing viable solutions to the stability issues typical of NIR QDs capped with pure organic ligand shells.
 Please wait while we load your content...
Please wait while we load your content...

