
Differential parsing of EGFR endocytic flux among parallel internalization pathways in lung cancer cells with EGFR-activating mutations†
Abstract
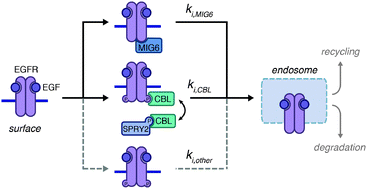
Due to the existence of parallel pathways for receptor endocytosis and their complexities, a quantitative understanding of receptor endocytosis in normal and pathological settings requires computational analysis. Here, we develop a mechanistic model of epidermal growth factor receptor (EGFR) endocytosis to determine the relative contributions of three parallel pathways: clathrin-dependent internalization mediated by mitogen-inducible gene 6 (MIG6), an endogenous EGFR kinase inhibitor that links EGFR to endocytic proteins; clathrin-dependent internalization mediated by the ubiquitin ligase CBL, which can be sequestered by the regulatory protein Sprouty2; or alternative pathways that may be non-clathrin mediated. We applied the model to interpret our previous measurements of EGFR endocytosis in lung cancer cells. Interestingly, our results suggest that MIG6 is responsible for at least as much wild-type EGFR internalization as CBL, indicating that a significant fraction of internalizing EGFR may be incapable of driving signaling. Model results also suggest that MIG6's endocytic function is reduced for the kinase-activated and internalization-impaired EGFR mutants found in some lung cancers. Analysis of Sprouty2 knockdown data indicates that Sprouty2 regulates EGFR endocytosis primarily by controlling EGFR expression, rather than by sequestering CBL, and supports the notion that CBL-mediated internalization is impaired for EGFR mutants. We further demonstrate that differences in internalization between wild-type and mutant EGFR cannot explain differences in EGF-mediated EGFR degradation without concomitant changes in EGFR recycling, which we previously quantified. This work provides new quantitative insights into EGFR trafficking in lung cancer and provides a framework for studying parallel endocytosis pathways for other receptors.
 Please wait while we load your content...
Please wait while we load your content...