
Using solution- and solid-state S K-edge X-ray absorption spectroscopy with density functional theory to evaluate M–S bonding for MS42− (M = Cr, Mo, W) dianions†
Abstract
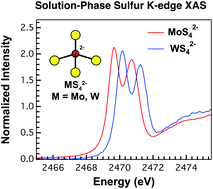
Herein, we have evaluated relative changes in M–S electronic structure and orbital mixing in Group 6 MS42− dianions using solid- and solution-phase S K-edge X-ray absorption spectroscopy (XAS; M = Mo, W), as well as density functional theory (DFT; M = Cr, Mo, W) and time-dependent density functional theory (TDDFT) calculations. To facilitate comparison with solution measurements (conducted in acetonitrile), theoretical models included gas-phase calculations as well as those that incorporated an acetonitrile dielectric, the latter of which provided better agreement with experiment. Two pre-edge features arising from S 1s → e* and t*2 electron excitations were observed in the S K-edge XAS spectra and were reasonably assigned as 1A1 → 1T2 transitions. For MoS42−, both solution-phase pre-edge peak intensities were consistent with results from the solid-state spectra. For WS42−, solution- and solid-state pre-edge peak intensities for transitions involving e* were equivalent, while transitions involving the t*2 orbitals were less intense in solution. Experimental and computational results have been presented in comparison to recent analyses of MO42− dianions, which allowed M–S and M–O orbital mixing to be evaluated as the principle quantum number (n) for the metal valence d orbitals increased (3d, 4d, 5d). Overall, the M–E (E = O, S) analyses revealed distinct trends in orbital mixing. For example, as the Group 6 triad was descended, e* (π*) orbital mixing remained constant in the M–S bonds, but increased appreciably for M–O interactions. For the t*2 orbitals (σ* + π*), mixing decreased slightly for M–S bonding and increased only slightly for the M–O interactions. These results suggested that the metal and ligand valence orbital energies and radial extensions delicately influenced the orbital compositions for isoelectronic ME42− (E = O, S) dianions.
 Please wait while we load your content...
Please wait while we load your content...

