
Addition of C–C and C–H bonds by pincer-iridium complexes: a combined experimental and computational study†‡
Abstract
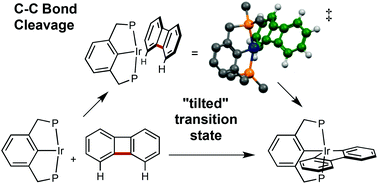
We report that pincer-ligated iridium complexes undergo oxidative addition of the strained C–C bond of biphenylene. The sterically crowded species (tBuPCP)Ir (RPCP = κ3-1,3-C6H3(CH2PR2)2) initially reacts with biphenylene to selectively add the C(1)–H bond, to give a relatively stable aryl hydride complex. Upon heating at 125 °C for 24 h, full conversion to the C–C addition product, (tBuPCP)Ir(2,2′-biphenyl), is observed. The much less crowded (iPrPCP)Ir undergoes relatively rapid C–C addition at room temperature. The large difference in the apparent barriers to C–C addition is notable in view of the fact that the addition products are not particularly crowded, since the planar biphenyl unit adopts an orientation perpendicular to the plane of the RPCP ligands. Based on DFT calculations the large difference in the barriers to C–C addition can be explained in terms of a “tilted” transition state. In the transition state the biphenylene cyclobutadiene core is calculated to be strongly tilted (ca. 50°–60°) relative to its orientation in the product in the plane perpendicular to that of the PCP ligand; this tilt results in very short, unfavorable, non-bonding contacts with the t-butyl groups in the case of the tBuPCP ligand. The conclusions of the biphenylene studies are applied to interpret computational results for cleavage of the unstrained C–C bond of biphenyl by (RPCP)Ir.
- This article is part of the themed collection: The Carbon–Metal Bond and C–H Metalation
 Please wait while we load your content...
Please wait while we load your content...

