
Computational evaluation of tris(carbene)borate donor properties in {NiNO}10 complexes†
Abstract
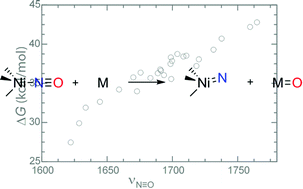
Electronic structure calculations are performed to characterise the structures, energies, and spectroscopic data for a series of four-coordinate tris(carbene)borate {NiNO}10 complexes. There is excellent agreement between the computational and experimental results for known complexes, allowing for structure–function relationships to be delineated. Calculations that provide insights into the synthetic accessibility of nickel(IV) nitrides by oxygen atom abstraction from these complexes are also reported.
 Please wait while we load your content...
Please wait while we load your content...

