
Combined DFT and experimental studies of C–C and C–X elimination reactions promoted by a chelating phosphine–alkene ligand: the key role of penta-coordinate PdII†
Abstract
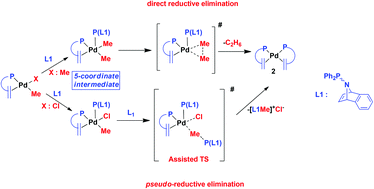
A combined computational and experimental study of the coordination chemistry of phosphine–alkene ligand L1 (N-diphenylphosphino-7-aza-benzobicyclo[2.2.1]hept-2-ene) with Pd0 and PdII is presented. Experimentally it is established that ligand L1 promotes direct alkyl–alkyl and indirect alkyl–halide reductive elimination from PdII species, affording the palladium(0) complex [Pd(κ2-P,C-L1)2] (2) in each case. The effectiveness of L1 in promoting these reactions is attributed to the initial formation of a penta-coordinate intermediate [PdMe(X)(κ1-P-L1)(κ2-P,C-L1)] (X = Me, Cl) coupled with the ease with which it transforms to 2. From computation, a lower activation barrier for C(sp3)–C(sp3) coupling and subsequent elimination has been computed for a stepwise associative pathway involving the initial formation of [PdMe2(κ1-P-L1)(κ2-P,C-L1)], compared to that computed for direct elimination from its parent, cis-[PdMe2(κ2-P,C-L1)]. Moreover, the C(sp3)–C(sp3) coupling reaction has been found to be primarily under thermodynamic control. It has also been demonstrated computationally that the methyl group of penta-coordinate [PdCl(Me)(κ1-P-L1)(κ2-P,C-L1)] is susceptible to nucleophilic attack by the phosphorus lone pair of a further equivalent of ligand L1, which proceeds through an SN2-like transition state. This initiates an unusual, indirect intermolecular reductive elimination process, resulting in the formation of equimolar quantities of the methyl phosphonium chloride salt of L1 and complex 2, in agreement with experimental observations. In contrast to the C(sp3)–C(sp3) coupling, computation shows that this indirect C(sp3)–Cl reductive elimination process is essentially under kinetic control.
- This article is part of the themed collection: Synergy between Experiment and Theory
 Please wait while we load your content...
Please wait while we load your content...

