
Accurate prediction of 195Pt NMR chemical shifts for a series of Pt(ii) and Pt(iv) antitumor agents by a non-relativistic DFT computational protocol†
Abstract
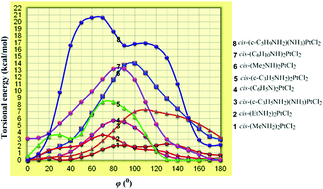
The GIAO-PBE0/SARC-ZORA(Pt)∪6-31+G(d)(E) (E = main group element) computational protocol without including relativistic and spin–orbit effects is offered here for the accurate prediction of the 195Pt NMR chemical shifts of a series of cis-(amine)2PtX2 (X = Cl, Br, I) anticancer agents (in total 42 complexes) and cis-diacetylbis(amine)platinum(II) complexes (in total 12) in solutions employing the Polarizable Continuum Model (PCM) solvation model, thus contributing to the difficult task of computation of 195Pt NMR. Calculations of the torsional energy curves along the diabatic (unrelaxed) rotation around the Pt–N bond of the cis-(amine)2PtX2 (X = Cl, Br, I) anticancer agents revealed the high sensitivity of the 195Pt NMR chemical shifts to conformational changes. The crucial effect of the conformational preferences on the electron density of the Pt central atom and consequently on the calculated δ195Pt chemical shifts was also corroborated by the excellent linear plots of δcalcd(195Pt) chemical shifts vs. the natural atomic charge QPt. Furthermore, for the accurate prediction of the 195Pt NMR chemical shifts of the cis-bis(amine)Pt(II) anticancer agents bearing carboxylato- as the leaving ligands (in total 8) and a series of octahedral Pt(IV) antitumor agents (in total 20 complexes) the non-relativistic GIAO-PBE0/SARC-ZORA(Pt)∪6-31+G(d)(E) computational protocol performs best in combination with the universal continuum solvation model based on solute electron density called SMD for aqueous solutions. Despite neglecting relativistic and spin orbit effects the agreement of the calculated δ195Pt chemical shifts with experimental values is surprising probably due to effective error compensation. Moreover, the observed solvent effects on the structural parameters of the complexes probably overcome the relativistic effects, and therefore the successful applicability of the non-relativistic GIAO-PBE0/SARC-ZORA(Pt)∪6-31+G(d)(E) computational protocol in producing reliable δcalcd(195Pt) chemical shifts could be understood. In a few cases (e.g. the dihydroxo Pt(IV) complexes) the higher deviations of the calculated from the experimental values of δ195Pt chemical shifts are probably due to the fact that the experimental assignments refer to a different composition of the complexes in solutions than that used in the calculations, and different hydrogen bonding and formation of dimeric species.
 Please wait while we load your content...
Please wait while we load your content...

