
First-principles study of borohydride adsorption properties on osmium nanoparticles and surfaces: understanding the effects of facets, size and local sites
Abstract
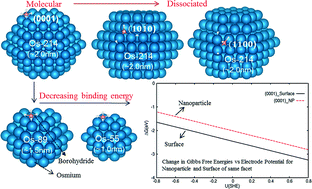
We present the first density functional theory investigation on the adsorption properties of borohydride on Os surfaces and nanoparticles with respect to the effects of facets, size and local sites. We found that the adsorption configuration and the binding energy significantly changed with respect to these factors. On Os surfaces, the most stable adsorbate configuration is molecular on (0001) but dissociated on (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) and (100). For the Os nanoparticles, the preferred configurations on the Os surface are preserved only on the counterpart (0001) and (100) planes and the binding energies are significantly larger due to the presence of vertices/edges. The difference in the structures between the (100) facet of the slab and of the nanoparticle is attributed to very different Os lateral distances. With respect to the nanoparticle size, the adsorbate structure is also preserved but the binding energy increases when the particle size is increased. At various electrode potentials, borohydride oxidative adsorption is less favorable on the ~2.0 nm Os nanoparticles as compared to the Os surface of the same facet due to enhanced solvation at the vertex sites of the former. Finally, using ab initio molecular dynamics, we found that these less coordinated sites of the Os nanoparticles reconstruct significantly with temperature, especially the smaller sized ones. Only when the nanoparticle size is ~2.0 nm do these reconstructions become minimal. These findings present fundamental borohydride adsorption information on nanoparticles and surface facets relevant for modeling/screening of suitable nanostructures of anode catalysts for direct borohydride fuel cells.
0) and (100). For the Os nanoparticles, the preferred configurations on the Os surface are preserved only on the counterpart (0001) and (100) planes and the binding energies are significantly larger due to the presence of vertices/edges. The difference in the structures between the (100) facet of the slab and of the nanoparticle is attributed to very different Os lateral distances. With respect to the nanoparticle size, the adsorbate structure is also preserved but the binding energy increases when the particle size is increased. At various electrode potentials, borohydride oxidative adsorption is less favorable on the ~2.0 nm Os nanoparticles as compared to the Os surface of the same facet due to enhanced solvation at the vertex sites of the former. Finally, using ab initio molecular dynamics, we found that these less coordinated sites of the Os nanoparticles reconstruct significantly with temperature, especially the smaller sized ones. Only when the nanoparticle size is ~2.0 nm do these reconstructions become minimal. These findings present fundamental borohydride adsorption information on nanoparticles and surface facets relevant for modeling/screening of suitable nanostructures of anode catalysts for direct borohydride fuel cells.
 Please wait while we load your content...
Please wait while we load your content...

