
Kinetic Monte Carlo simulations of heterogeneously catalyzed oxidation reactions†
Abstract
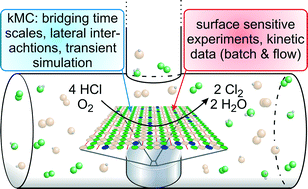
In this perspective, we focus on the catalyzed oxidation of CO and HCl over the model catalyst RuO2(110) and how the kinetics of these reactions can be modeled by kinetic Monte Carlo (kMC) simulations. Assuming the reaction mechanism is known, the critical parameters entering the kMC simulations include the activation and adsorption energies as well as interaction energies between the adsorbed species and the diffusion barriers. This input parameter set can either be determined by using dedicated coadsorption experiments or by calculations from electronic structure theory. A critical comparison of kMC results with on-line kinetic and in situ spectroscopic experiments enables the assessment of a proposed reaction mechanism. Transient rather than steady state experiments are of particular importance for this purpose. Only the inclusion of lateral interactions among the reaction intermediates allows for the determination of an apparent activation energy which is consistent with the experiment. For the case of CO oxidation over RuO2(110), we compare the results of kMC with those based on the mean field approach, the standard method of microkinetic modeling. It turns out that under realistic reaction conditions for the CO oxidation over RuO2(110) both methods can equally well describe experimental kinetic data if lateral repulsion is included in the model. However, if one-dimensional confinement is encountered such as with the HCl oxidation reaction over RuO2(110), then kMC is the preferred method for microkinetic modeling.
 Please wait while we load your content...
Please wait while we load your content...

