
Abstraction and addition kinetics of C2H radicals with CH4, C2H6, C3H8, C2H4, and C3H6: CVT/SCT/ISPE and hybrid meta-DFT methods†
Abstract
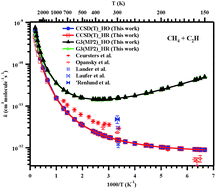
Rate coefficients for the reactions of C2H radicals with methane (k1), ethane (k2), propane (k3), ethylene (k4), and propylene (k5) were computed using canonical variational transition state theory (CVT) coupled with hybrid-meta density functional theory (DFT) over a wide range of temperatures from 150 to 5000 K. The quantum chemical tunneling effect was corrected by the small curvature tunneling (SCT) method. The dynamic calculations are performed using the variational transition state theory (VTST) with the interpolated single-point energies (ISPE) method at the CCSD(T)/cc-pVTZ//M06-2X/6-31+G(d,p) level of theory. Intrinsic reaction coordinate (IRC) calculations were performed to verify that the transition states are connected to the reactants and products. The rate coefficients obtained over the studied temperature range yield the following Arrhenius expressions (cm3 molecule−1 s−1): k1 = 4.69 × 10−19T2.44 exp[331/T], k2 = 4.29 × 10−17T2.11 exp[432/T], k3 = 4.81 × 10−17T1.98 exp[697/T], k4 = 7.54 × 10−21T2.96 exp[1942/T], and k5 = 8.04 × 10−23T3.44 exp[3011/T] cm3 molecule−1 s−1. Branching ratio calculation for the reactions of C2H radicals with ethylene and propylene shows that the abstraction reactions are not important at lower temperatures. However, as the temperature increases, abstraction reactions become more important.
 Please wait while we load your content...
Please wait while we load your content...

