
Direct vs. indirect pathway for nitrobenzene reduction reaction on a Ni catalyst surface: a density functional study
Abstract
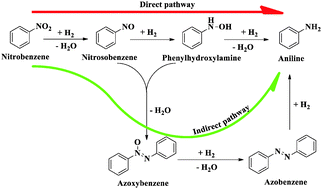
Density functional theory (DFT) calculations are performed to understand and address the previous experimental results that showed the reduction of nitrobenzene to aniline prefers direct over indirect reaction pathways irrespective of the catalyst surface. Nitrobenzene to aniline conversion occurs via the hydroxyl amine intermediate (direct pathway) or via the azoxybenzene intermediate (indirect pathway). Through our computational study we calculated the spin polarized and dispersion corrected reaction energies and activation barriers corresponding to various reaction pathways for the reduction of nitrobenzene to aniline over a Ni catalyst surface. The adsorption behaviour of the substrate, nitrobenzene, on the catalyst surface was also considered and the energetically most preferable structural orientation was elucidated. Our study indicates that the parallel adsorption behaviour of the molecules over a catalyst surface is preferable over vertical adsorption behaviour. Based on the reaction energies and activation barrier of the various elementary steps involved in direct or indirect reaction pathways, we find that the direct reduction pathway of nitrobenzene over the Ni(111) catalyst surface is more favourable than the indirect reaction pathway.
 Please wait while we load your content...
Please wait while we load your content...

