
A combined MD/QM and experimental exploration of conformational richness in branched oligothiophenes†
Abstract
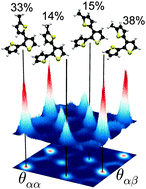
Infrared (IR) absorption and vibrational Raman spectra of a family of branched oligothiophenes have been determined experimentally as well as theoretically. The molecular spectra have been compared to those of the linear analogues, with identification made of spectral features due to structural properties that are valued in organic solar cell applications. The theoretical spectra have been obtained through a newly developed method in which individual conformer spectra, calculated at the time-dependent DFT level in this work, are weighted by statistics extracted from classical molecular dynamics trajectories. The agreement with experiment for the resulting averaged spectra is at least as good as, and often better than, what is observed for Boltzmann-weighted spectra. As the weights are available before the costly step of spectrum calculation, the method has the additional advantage of enabling efficient approximations. For simulating the molecular dynamics of the studied α,β-linked thiophenes and 2-methylthiophenes, high quality parameters have been derived for the CHARMM force field. Furthermore, the temperature dependence of the IR and Raman spectra has been investigated, both experimentally and theoretically.
 Please wait while we load your content...
Please wait while we load your content...

