
Mapping the affinity of aluminum(iii) for biophosphates: interaction mode and binding affinity in 1 : 1 complexes†
Abstract
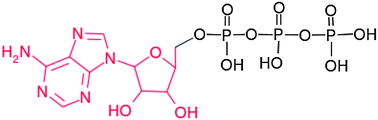
The interaction of aluminum with biomolecular building blocks is a topic of interest as a first step to understand the potential toxic effects of aluminum in biosystems. Among the different molecules that aluminum can bind in a biological environment, phosphates are the most likely ones, due to their negatively charged nature. In the present paper, we combined DFT quantum mechanical calculations with the implicit solvent effect in order to characterize the interaction of Al(III) with these molecules. An extended set composed of a total of 59 structures was investigated, which includes various types of phosphates (monoester, diester, triester-phosphates) and various phosphate units (mono-, di- and tri-phosphate), considering various charge and protonation states, and different binding modes. The goal is to unveil the preferential interaction mode of Al(III) with phosphates in 1 : 1 complexes. Our results reveal that Al(III) prefers to form dicoordinated complexes with two phosphates, in which the interaction with each of the phosphates is of monodentate character. Our results also suggest a high affinity for binding basic phosphate groups, pointing to ATP, phosphorylated peptides, and basic diphosphates (such as 2,3-DPG) as strong aluminum chelators.
 Please wait while we load your content...
Please wait while we load your content...

