
Insights into the interactions of CO2 with amines: a DFT benchmark study†
Abstract
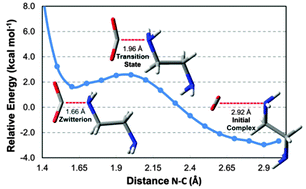
The interaction between CO2 and 1,2-diaminoethane was computed using pure and hybrid density functionals. The CAM-B3LYP and wB97X-D functionals using a triple-ζ basis set that includes diffuse and polarization functions are the best functionals for calculating the relative energies of the zwitterion intermediate compared to a coupled-cluster with a single, double and non-iterative triple excitation (CCSD(T)) approach extrapolated to a complete basis set limit. With the two functionals and the triple-ζ basis set, the zwitterion is 1.70 kcal mol−1 less stable than the reactants, and close to 1.63 kcal mol−1 computed using the CCSD(T) approach. The inclusion of vibrational and thermal corrections and of entropic effects increases the relative energy of the zwitterion to 14.7 kcal mol−1. Bending of the CO2 geometry increases its acidity due to a 1.09 eV reduction in the LUMO energy. Calculation of the CO2 interaction energy with a set of amines revealed that the interaction energies show a high correlation with their basicities, with the stronger bases stabilizing the zwitterion. For the most basic amine computed (3,4,6,7,8,9-hexahydro-2H-pyrimido[1,2-a]pyrimidine), the Gibbs free energy of the zwitterion is 15.8 kcal mol−1 lower than the reactants. Therefore, for this highly basic amine, the zwitterion may have a longer lifetime, in contrast to 2-aminoethanol (MEA), where it is only a transient species.
 Please wait while we load your content...
Please wait while we load your content...

