
A quantum mechanical study of water adsorption on the (110) surfaces of rutile SnO2 and TiO2: investigating the effects of intermolecular interactions using hybrid-exchange density functional theory
Abstract
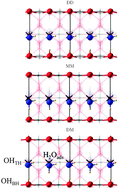
Periodic hybrid-exchange density functional theory calculations are used to explore the first layer of water at model oxide surfaces, which is an important step for understanding the photocatalytic reactions involved in solar water splitting. By comparing the structure and properties of SnO2(110) and TiO2(110) surfaces in contact with water, the effects of structural and electronic differences on the water chemistry are examined. The dissociative adsorption mode at low coverage (1/7 ML) up to monolayer coverage (1 ML) on both SnO2 and TiO2(110) surfaces is analysed. To investigate further the intermolecular interactions between adjacent adsorbates, monolayer adsorption on each surface is explored in terms of binding energies and bond lengths. Analysis of the water adsorption geometry and energetics shows that the relative stability of water adsorption on SnO2(110) is governed largely by the strength of the chemisorption and hydrogen bonds at the surface of the adsorbate–substrate system. However on TiO2(110), a more complicated scenario of the first layer of water on its surface arises in which there is an interplay between chemisorption, hydrogen bonding and adsorbate-induced atomic displacements in the surface. Furthermore the projected density of states of each surface in contact with a mixture of adsorbed water molecules and adsorbed hydroxyls is presented and sheds some light on the nature of the crystalline chemical bonds as well as on why adsorbed water has often been reported to be unstable on rutile SnO2(110).
- This article is part of the themed collection: High performance computing in the chemistry of materials
 Please wait while we load your content...
Please wait while we load your content...

