
Microsecond molecular dynamics simulation of guanidinium chloride induced unfolding of ubiquitin†
Abstract
An all atom molecular dynamics simulation was used to explore the atomic detail mechanism of guanidinium induced unfolding of the protein ubiquitin. Ubiquitin unfolds through pre-unfolded (intermediate) states, i.e. guanidinium induced unfolding of ubiquitin appears to be a multi-step process, and loss of hydrophobic contacts of C-terminal residues is crucial for ubiquitin unfolding. Free-energy landscapes show that barrier separation between folded and unfolded basins is ∼5.0 kcal mol−1, and both the basins are of comparable energy. It was observed that guanidinium ions interact directly with ubiquitin. Favorable electrostatic interaction is the main driving force for such accumulation of guanidinium ions near protein, but van der Waals energy also contributes. RDF plots show that accumulation of guanidinium ions near specific residues is the main cause for destabilization of intra-residue interactions crucial to maintain the three-dimensional fold of the protein. One salt-bridge interaction between Lys11 and Glu34 appears to be important to maintain the crystal structure of ubiquitin and this salt-bridge can map the unfolding process of ubiquitin.
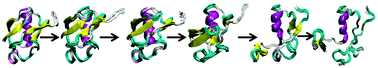
 Please wait while we load your content...
Please wait while we load your content...

