
Ab initio theoretical investigation of beryllium and beryllium hydride nanoparticles and nanocrystals with implications for the corresponding infinite systems
Abstract
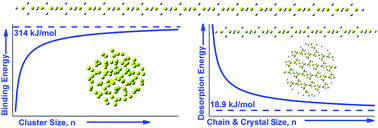
With the initial motivation of optimizing hydrogen storage in beryllium nanocrystals, we have thoroughly and systematically studied the structural, cohesive, and electronic properties of Ben and BenHxn (n = 2–160, x = 0.1–2.4) nanoparticles as a function of both size (n) and hydrogen content (x), using density functional theory with a properly selected meta-hybrid functional and high level coupled cluster CCSD(T) theory for comparison. We have calculated the binding energies of Ben, BenHxn and [BeH2]n nanoparticles for a large range of n values. In the limit n → ∞, we have obtained the experimental binding energy of a Be crystal (3.32 eV) with unexpectedly very good agreement (3.26 ± 0.06 eV), and a predicted value of 7.85 eV ± 0.02 eV for the binding energy of the [BeH2]∞ infinite system. We also predict that the majority of the lowest energy stoichiometric BenH2n nanoparticles are chains or chain-like structures. The tendency towards chain stabilization of BenHxn nanoparticles increases, as x approaches the stoichiometric value x = 2, leading for large values of n, as n → ∞, to polymeric forms of bulk BeH2, which in the past have been considered as the leading forms of solid BeH2. For such 1-dimensional forms of [BeH2]n we have obtained and verified that the binding energy varies exactly proportionally to n−1. The extrapolated desorption energy for such polymeric forms of solid BeH2 is found to be 19 ± 3 kJ mol−1 in juxtaposition to the experimental value of 19 kJ mol−1 for solid BeH2, suggesting that the difference ΔE in cohesive energy between the orthorhombic and polymeric form is very small (ΔE ≈ 3 kJ mol−1). This is in full accord with the early discrepancies in the literature in determining and distinguishing the real crystal structure of solid BeH2.
 Please wait while we load your content...
Please wait while we load your content...

