
A computational study on the complexation of Np(v) with N,N,N′,N′-tetramethyl-3-oxa-glutaramide (TMOGA) and its carboxylate analogs†
Abstract
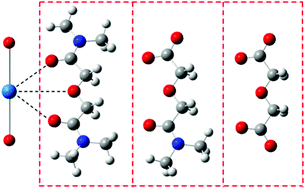
Density functional theory has been used to study the geometries and relative stabilities of the complexes of NpO2+ with the title compounds (L), including TMOGA, deprotonated N,N′-dimethyl-3-oxa-glutaramic acid (DMOGA) and their deprotonated oxydiacetic analog (ODA). Our calculations suggest that the complexes where the ligands appear as tridentate chelators are more stable than as bidentate ones, and the substitution of the amide group by carboxylate favors the formation of the complexes. Thermodynamically the 1 : 2 complex (Np–L2) is more favorable than the 1 : 1 complex (Np–L) in the cases of TMOGA and DMOGA, but not for the ODA anion. Taking into account the solvation effect of water, the 1 : 2 complex is more favorable than the 1 : 1 complex for all of the three ligands, though the reaction enthalpy decreases compared to that in the gas phase, and the formation of Np–(TMOGA)2 from Np–TMOGA is roughly a thermal neutral process. The strength of the Np![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond is weakened upon the coordination of ligands to Np(V) and the increase of the negative charge on the ligand (−1e for deprotonated DMOGA and −2e for deprotonated ODA). The Quantum Theory of Atoms-in-Molecules (QTAIM) was used here to analyze the bonding mode of NpO2+–Lx (x = 1, 2) and to compare the bond order data.
O bond is weakened upon the coordination of ligands to Np(V) and the increase of the negative charge on the ligand (−1e for deprotonated DMOGA and −2e for deprotonated ODA). The Quantum Theory of Atoms-in-Molecules (QTAIM) was used here to analyze the bonding mode of NpO2+–Lx (x = 1, 2) and to compare the bond order data.
 Please wait while we load your content...
Please wait while we load your content...

