
Performance of the MM/GBSA scoring using a binding site hydrogen bond network-based frame selection: the protein kinase case†
Abstract
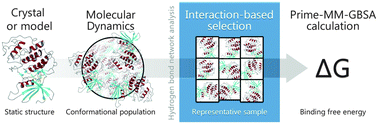
A conformational selection method, based on hydrogen bond (Hbond) network analysis, has been designed in order to rationalize the configurations sampled using molecular dynamics (MD), which are commonly used in the estimation of the relative binding free energy of ligands to macromolecules through the MM/GBSA or MM/PBSA method. This approach makes use of protein–ligand complexes obtained from X-ray crystallographic data, as well as from molecular docking calculations. The combination of several computational approaches, like long MD simulations on protein–ligand complexes, Hbond network-based selection by scripting techniques and finally MM/GBSA, provides better statistical correlations against experimental binding data than previous similar reported studies. This approach has been successfully applied in the ranking of several protein kinase inhibitors (CDK2, Aurora A and p38), which present both diverse and related chemical structures.
 Please wait while we load your content...
Please wait while we load your content...

