
Optimizing porphyrins for dye sensitized solar cells using large-scale ab initio calculations†
Abstract
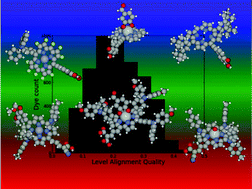
In the search for sustainable energy sources, dye sensitized solar cells (DSSCs) represent an attractive solution due to their low cost, relatively high efficiencies, and flexible design. Porphyrin-based dyes are characterized by strong absorption in the visible part of the spectrum and easy customization allowing their electronic properties to be controlled by structural variations. Here we present a computational screening study of more than 5000 porphyrin-based dyes obtained by modifying the porphyrin backbone (metal center and axial ligands), substituting hydrogen by fluorine, and adding different side and anchoring groups. Based on the calculated frontier orbital energies and optical gaps we quantify the energy level alignment with the TiO2 conduction band and different redox mediators. An analysis of the energy level–structure relationship reveals a significant structural diversity among the dyes with the highest level alignment quality, demonstrating the large degree of flexibility in porphyrin dye design. As a specific example of dye optimization, we show that the level alignment of the high efficiency record dye YD2-o-C8 [Yella et al., Science, 2011, 334, 629–634] can be significantly improved by modest structural variations. All the presented data have been stored in a publicly available database.
 Please wait while we load your content...
Please wait while we load your content...

