
On the molecular mechanism of non-radiative decay of nitrobenzene and the unforeseen challenges this simple molecule holds for electronic structure theory†
Abstract
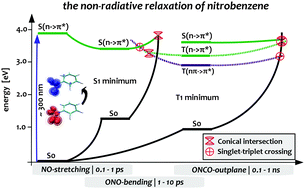
In this work, we present a complete mechanistic picture of the non-radiative decay of the mono-substituted aromatic compound nitrobenzene from the bright singlet state to the electronic ground state. This mechanism involves internal conversion (IC) and inter-system crossing (ISC) along three dominating internal coordinates of the nitro group and consistently explains the experimental findings. Relaxation from the lowest triplet state via ISC occurs along the out-of-plane bending coordinate of the nitro group, while initial IC as well as ultrafast ISC into the triplet manifold take place along symmetric NO stretching and ONO bending modes that have not been considered yet. The proposed mechanism is based on high-level single- and multi-reference electronic structure calculations employing ADC3, MOM-CCSD(T), EOM-CCSD, DFT/MRCI and CAS-SCF/NEVPT2 levels of theory, which is, as we will demonstrate, absolutely necessary to assure a reliable and sufficiently accurate theoretical description of nitrobenzene. The need for third-order methods will be traced back to the large double-excitation character of about 50% of the second excited singlet state of nitrobenzene. As a result, second-order methods like approximate coupled-cluster of second order (CC2) and partially even (EOM-)CCSD yield a qualitatively wrong picture of the excited states. Surprisingly, already the description of the ground state geometries is problematic at the CC2 and partially also CCSD level of theory.
 Please wait while we load your content...
Please wait while we load your content...

