
Substituted diphenyl butadiynes: a computational study of geometries and electronic transitions using DFT/TD-DFT†
Abstract
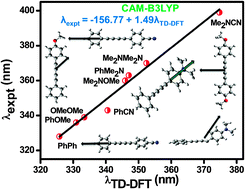
This work is aimed at theoretical understanding of electronic absorption and emission energies of a series of substituted diphenyl butadiynes through an assessment of several TDDFT functionals and a detailed study of solvent effects on their ground and excited state structures and properties. Out of a series of functionals examined, the coulomb attenuated DFT functional CAM-B3LYP is found to be most successful in predicting charge transfer absorption and emission energies of such derivatives. However, TDDFT potential energy surfaces obtained from hybrid functionals such as B3LYP and PBE0 are found to give a good description of the stability of locally excited (LE) and intramolecular charge transfer (ICT) states as a function of torsional angle, for the butadiynyl fluorophores. Interesting structural variations are observed in the ground and excited state optimized geometries of the fluorophores. The ICT emission of the butadiynyl fluorophores is observed to originate from the twisted state where the two phenyl rings in the diphenyl butadiyne get twisted around the butadiyne moiety. A bending of the butadiyne moiety is noted for some of the butadiynyl derivatives in the ICT emissive state. In addition, the direction of absorption and emission transition dipole moment vectors of the butadiynyl fluorophores is found to depend on the nature of substituents present at the periphery of the diphenyl butadiyne moiety.
 Please wait while we load your content...
Please wait while we load your content...

