
An interatomic potential for saturated hydrocarbons based on the modified embedded-atom method
Abstract
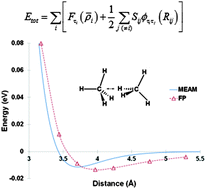
In this work, we developed an interatomic potential for saturated hydrocarbons using the modified embedded-atom method (MEAM), a reactive semi-empirical many-body potential based on density functional theory and pair potentials. We parameterized the potential by fitting to a large experimental and first-principles (FP) database consisting of (1) bond distances, bond angles, and atomization energies at 0 K of a homologous series of alkanes and their select isomers from methane to n-octane, (2) the potential energy curves of H2, CH, and C2 diatomics, (3) the potential energy curves of hydrogen, methane, ethane, and propane dimers, i.e., (H2)2, (CH4)2, (C2H6)2, and (C3H8)2, respectively, and (4) pressure–volume–temperature (PVT) data of a dense high-pressure methane system with the density of 0.5534 g cc−1. We compared the atomization energies and geometries of a range of linear alkanes, cycloalkanes, and free radicals calculated from the MEAM potential to those calculated by other commonly used reactive potentials for hydrocarbons, i.e., second-generation reactive empirical bond order (REBO) and reactive force field (ReaxFF). MEAM reproduced the experimental and/or FP data with accuracy comparable to or better than REBO or ReaxFF. The experimental PVT data for a relatively large series of methane, ethane, propane, and butane systems with different densities were predicted reasonably well by the MEAM potential. Although the MEAM formalism has been applied to atomic systems with predominantly metallic bonding in the past, the current work demonstrates the promising extension of the MEAM potential to covalently bonded molecular systems, specifically saturated hydrocarbons and saturated hydrocarbon-based polymers. The MEAM potential has already been parameterized for a large number of metallic unary, binary, ternary, carbide, nitride, and hydride systems, and extending it to saturated hydrocarbons provides a reliable and transferable potential for atomistic/molecular studies of complex material phenomena involving hydrocarbon–metal or polymer–metal interfaces, polymer–metal nanocomposites, fracture and failure in hydrocarbon-based polymers, etc. The latter is especially true since MEAM is a reactive potential that allows for dynamic bond formation and bond breaking during simulation. Our results show that MEAM predicts the energetics of two major chemical reactions for saturated hydrocarbons, i.e., breaking a C–C and a C–H bond, reasonably well. However, the current parameterization does not accurately reproduce the energetics and structures of unsaturated hydrocarbons and, therefore, should not be applied to such systems.
 Please wait while we load your content...
Please wait while we load your content...

