
Sensitivity of local hydration behaviour and conformational preferences of peptides to choice of water model
Abstract
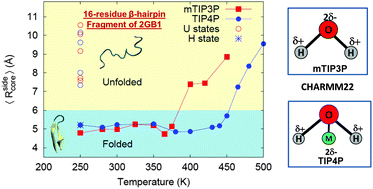
Hydration of the 16-residue β-hairpin fragment of the 2GB1 protein in the folded and unfolded ensembles is studied with mTIP3P and TIP4P solvent models using the CHARMM22 protein force-field. mTIP3P is a three-site water model which is used for parameterization of the CHARMM force-field and is known to exhibit liquid-state anomalies of water at temperatures about 80 K lower than the experimental temperature. TIP4P is a four-site water model which gives a better description of the experimental phase diagram and liquid-state anomalies of water. At a temperature of 250 K, where the folded ensemble of the peptide is stable and the unfolded ensemble is metastable, secondary structure metrics are much more sensitive to the choice of solvent model in the unfolded, rather than folded, ensemble. In particular, mean values as well as variation in the positional root mean square displacements (RMSD) and configurational entropy are greater in mTIP3P compared to the TIP4P solvent. The peptide structure is relatively more compact in the TIP4P solvent, which supports unfolded as well as hydrophobic core states. In terms of average local order and binding energy of the water surrounding the peptide, strong deviations from bulk behaviour are restricted to the first hydration shell and differences between the folded and unfolded ensembles in the two solvents are small. The strong coupling between the solvent and the peptide is demonstrated, however, by the dependence of the unfolding temperature on the water model (400 K in mTIP3P and 465 K in TIP4P) and the qualitatively different temperature dependence of the hydration layer occupancy signalling the unfolding transition in the two solvents. A residue-wise decomposition of different contributions to the configurational energy indicates that the TIP4P solvent shows far greater variation in the interaction with charged sidegroups of amino acid residues than the mTIP3P solvent. The implications of sequence-dependent sensitivity of peptide secondary structures to the choice of water models for simulating folding–unfolding equilibria and free energy landscapes are discussed.
- This article is part of the themed collection: PCCP’s 15th anniversary
 Please wait while we load your content...
Please wait while we load your content...

