
Effects of hydrogen bonding interactions on the redox potential and molecular vibrations of plastoquinone as studied using density functional theory calculations†
Abstract
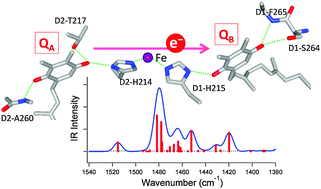
The effects of H-bonding on the redox potential and molecular vibrations of plastoquinone (PQ) that functions as a primary and a secondary quinone electron acceptor (QA and QB, respectively) in photosystem II (PSII) in plants and cyanobacteria were investigated using density functional theory calculations. Calculations were performed on the neutral and semiquinone anion forms of PQ and its H-bonded complexes, which form H-bonds with water molecules, or using amino acid models mimicking the interactions of QA and QB. The calculated redox potential (Eo) of PQ showed a linear relationship with the number of H-bonds, and the Eo increased by +100–200 mV with the addition of one H-bond. Vibrational analysis of the model PQ complexes showed that the CO stretching vibrations of neutral PQ are sensitive to the number and symmetry of H-bonding interactions, providing criteria to determine the H-bonding structure. Although no specific trend in the H-bonding dependency was found for anionic PQ, complex spectral features in the CO stretching region due to significant couplings with other PQ vibrations and the vibrations of H-bonding amino acids are useful monitors of the change in the H-bonding structure of anionic PQ in proteins. The calculated Eo values and infrared spectra of the QA and QB models are consistent with the view that one additional H-bond to QB from D1-Ser264 largely contributes to the redox potential gap between QA and QB in PSII.
- This article is part of the themed collection: Photosynthesis: From Natural to Artificial
 Please wait while we load your content...
Please wait while we load your content...

