
On the inter-ring torsion potential of regioregular P3HT: a first principles reexamination with explicit side chains†
Abstract
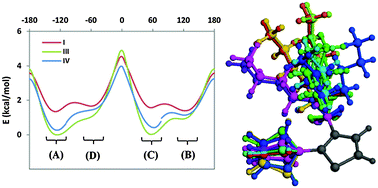
Poly(3-alkylthiophene) is a family of conjugated semicrystalline polymers for organic electronic applications. Crucial for the fine-tuning of such systems is a detailed understanding of the correlation between molecular structure/morphology and electronic properties. However, a series of a priori assumptions is commonly made in order to deduce macromolecular-scale geometric and energetic features from those of rather small homologous molecular systems. Alkyl side chains are routinely shortened (if not systematically removed) during such high-accuracy ab initio calculations in order to reduce their conformational space. We will show through first principles calculations on a monosubstituted bithiophene molecule how a full-length alkyl fragment can influence both side chain energetics and backbone flexibility in alkylthiophene-based polymers and copolymers. Folded side chains, characterized by a gauche arrangement of the second torsion angle from the ring, are found to be substantially favoured over extended ones, thanks to a network of CH–π hydrogen-bond-like interactions with both aromatic rings. Trans-planar (conjugated) arrangements of limit-ordered crystalline models, and cisoid sequences suitable for the investigation of chain-folding phenomena, are also discussed in detail.
 Please wait while we load your content...
Please wait while we load your content...

