
Coupled-cluster calculations of the lowest 0–0 bands of the electronic excitation spectrum of naphthalene†
Abstract
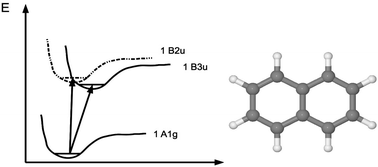
Approximate singles and doubles coupled-cluster (CC2) calculations have been carried out for the 0–0 bands of the 1Ag → 1B3u and 1Ag → 1B2u transitions of naphthalene. The vertical excitation energies calculated for the 16 lowest excited singlet states have also been calculated using a sequence of large basis sets. The CC2 excitation energies extrapolated to the basis-set limit are in rather good agreement with values recently calculated at the multiconfiguration second-order perturbation theory (CASPT2) and at the singles, doubles and approximate triples (CC3) levels. Best values for the vertical excitation energies and the 0–0 transition energies have been obtained by adding higher-order correlation contributions to the basis-set extrapolated CC2 energies. For some of the states, the best estimated vertical excitation energies in this work deviate up to 0.3 eV from the previously best estimated energies, because larger basis sets have been employed in this study. The calculations of the 0–0 transitions show the importance of considering vibrational effects when aiming at reliable comparisons of calculated and measured excitation energies for assessing the accuracy of employed computational methods. Calculations at the density functional theory (DFT) level using Becke's three-parameter functional (B3LYP) yield less accurate excitation energies. At the B3LYP level, the two lowest states appear in reverse order with a deviation of 0.50 eV from experiment for the 0–0 transition energy of the 1B3u state.
- This article is part of the themed collection: PCCP’s 15th anniversary
 Please wait while we load your content...
Please wait while we load your content...

