
Ab initio ORTEP drawings: a case study of N-based molecular crystals with different chemical nature
Abstract
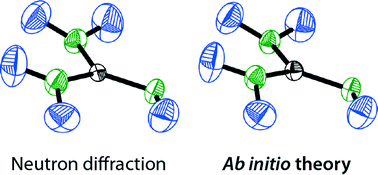
The thermal motion of atoms and functional groups is a key characteristic of any molecular crystal, and such motion derived from scattering experiments is conveniently visualised by means of thermal ellipsoids (the famous “ORTEP” drawings). Unfortunately, it is often impossible to obtain the underlying anisotropic displacement parameters (ADPs) for hydrogen atoms, due to their low X-ray scattering power, and sometimes no ADPs can be refined at all even for heavier atoms. In these cases, it would seem advantageous to estimate ADPs by first-principles techniques, and indeed such ab initio ORTEP plots have become available very recently. Here, we test this young method for a representative set of hydrogen-bonded molecular crystals: first, we study urea (CON2H4) as a well-known benchmark, then, its all-nitrogen analogue guanidine (CN3H5); finally, we move on to rubidium guanidinate (RbCN3H4) as a specimen with pronounced ionic interactions. For all three systems, ADPs have been obtained from density-functional theory (DFT) based phonon computations using the PHONOPY software. The results are compared with neutron-diffraction data as the experimental “benchmark” in this regard, and a critical discussion of experimental aspects is given. We observe excellent agreement between experiment and theory for the hydrogen-bonded systems urea and guanidine at low temperature, whereas high-temperature data for guanidine deviate visibly, and the more salt-like RbCN3H4 may suffer from a less-than-ideal description even at 12 K. Both are discussed in depth as there are possible solutions and directions for further research. Generally, the present results shine a favourable light on a future, more routine application of combined experimental/theoretical approaches in chemical crystallography.
 Please wait while we load your content...
Please wait while we load your content...

