
Metal control of selectivity in acetate-assisted C–H bond activation: an experimental and computational study of heterocyclic, vinylic and phenylic C(sp2)–H bonds at Ir and Rh†
Abstract
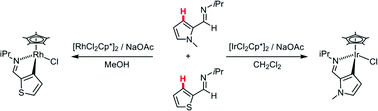
Acetate-assisted C(sp2)–H bond activation at [MCl2Cp*]2 (M = Ir, Rh) has been studied for a series of N-alkyl imines, iPrN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHR, (R = N-methyl-2-pyrrolyl, H-L1; 2-furanyl, H-L2; 2-thiophenyl, H-L3a; C2H2Ph, H-L4; and Ph, H-L5) as well as phenylpyridine (H-L6) by both experimental and computational means. Competition experiments reveal significant variation in the relative reactivity of these substrates and highlight changes in selectivity between Ir (H-L4 ≈ H-L2 < H-L3a ≈ H-L5 < H-L1 ≈ H-L6) and Rh (H-L2 ≈ H-L1 < H-L3a ≈ H-L4 < H-L5 < H-L6). Comparison of H-L3a with its N-xylyl analogue, H-L3b, gives a further case of metal-based selectivity, H-L3a being more reactive at Ir, while H-L3b is preferred at Rh. H/D exchange experiments suggest that the selectivity of C–H activation at Ir is determined by kinetic factors while that at Rh is determined by the product thermodynamic stability. This is confirmed by computational studies which also successfully model the order of substrate reactivity seen experimentally at each metal. To achieve the good level of agreement between experiment and computation required the inclusion of dispersion effects, use of large basis sets and an appropriate solvent correction.
CHR, (R = N-methyl-2-pyrrolyl, H-L1; 2-furanyl, H-L2; 2-thiophenyl, H-L3a; C2H2Ph, H-L4; and Ph, H-L5) as well as phenylpyridine (H-L6) by both experimental and computational means. Competition experiments reveal significant variation in the relative reactivity of these substrates and highlight changes in selectivity between Ir (H-L4 ≈ H-L2 < H-L3a ≈ H-L5 < H-L1 ≈ H-L6) and Rh (H-L2 ≈ H-L1 < H-L3a ≈ H-L4 < H-L5 < H-L6). Comparison of H-L3a with its N-xylyl analogue, H-L3b, gives a further case of metal-based selectivity, H-L3a being more reactive at Ir, while H-L3b is preferred at Rh. H/D exchange experiments suggest that the selectivity of C–H activation at Ir is determined by kinetic factors while that at Rh is determined by the product thermodynamic stability. This is confirmed by computational studies which also successfully model the order of substrate reactivity seen experimentally at each metal. To achieve the good level of agreement between experiment and computation required the inclusion of dispersion effects, use of large basis sets and an appropriate solvent correction.
 Please wait while we load your content...
Please wait while we load your content...

