
An ultra-stable oxoiron(iv) complex and its blue conjugate base†
Abstract
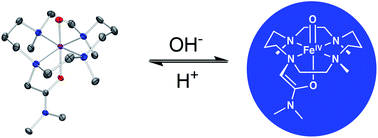
Treatment of [FeII(L)](OTf)2 (4), (where L = 1,4,8-Me3cyclam-11-CH2C(O)NMe2) with iodosylbenzene yielded the corresponding S = 1 oxoiron(IV) complex [FeIV(O)(L)](OTf)2 (5) in nearly quantitative yield. The remarkably high stability of 5 (t1/2 ≈ 5 days at 25 °C) facilitated its characterization by X-ray crystallography and a raft of spectroscopic techniques. Treatment of 5 with strong base was found to generate a distinct, significantly less stable S = 1 oxoiron(IV) complex, 6 (t1/2 ∼ 1.5 h at 0 °C), which could be converted back to 5 by addition of a strong acid; these observations indicate that 5 and 6 represent a conjugate acid–base pair. That 6 can be formulated as [FeIV(O)(L–H)](OTf) was further supported by ESI mass spectrometry, spectroscopic and electrochemical studies, and DFT calculations. The close structural similarity of 5 and 6 provided a unique opportunity to probe the influence of the donor trans to the FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O unit upon its reactivity in H-atom transfer (HAT) and O-atom transfer (OAT), and 5 was found to display greater reactivity than 6 in both OAT and HAT. While the greater OAT reactivity of 5 is expected on the basis of its higher redox potential, its higher HAT reactivity does not follow the anti-electrophilic trend reported for a series of [FeIV(O)(TMC)(X)] complexes (TMC = tetramethylcyclam) and thus appears to be inconsistent with the two-state reactivity rationale that is the prevailing explanation for the relative facility of oxoiron(IV) complexes to undergo HAT.
O unit upon its reactivity in H-atom transfer (HAT) and O-atom transfer (OAT), and 5 was found to display greater reactivity than 6 in both OAT and HAT. While the greater OAT reactivity of 5 is expected on the basis of its higher redox potential, its higher HAT reactivity does not follow the anti-electrophilic trend reported for a series of [FeIV(O)(TMC)(X)] complexes (TMC = tetramethylcyclam) and thus appears to be inconsistent with the two-state reactivity rationale that is the prevailing explanation for the relative facility of oxoiron(IV) complexes to undergo HAT.
 Please wait while we load your content...
Please wait while we load your content...

