
Tunable mechano-responsive organogels by ring-opening copolymerizations of N-carboxyanhydrides†
Abstract
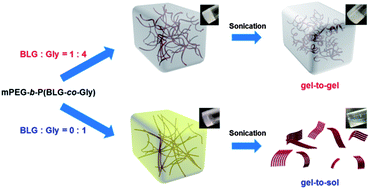
The simple copolymerization of N-carboxyanhydride (NCA) monomers is utilized to generate copolypeptides having a combination of α-helix and β-sheet sub-structures that, when grown from a solvophilic synthetic polymer block segment, are capable of driving mechano-responsive supramolecular sol-to-gel-to-sol and sol-to-gel-to-gel transitions reversibly, which allow also for injection-based processing and self-healing behaviors. A new type of polypeptide-based organogelator, methoxy poly(ethylene glycol)-block-poly(γ-benzyl-L-glutamate-co-glycine) (mPEG-b-P(BLG-co-Gly)), is facilely synthesized by statistical ring-opening copolymerizations (ROPs) of γ-benzyl-L-glutamate (BLG) and glycine (Gly) NCAs initiated by mPEG-amine. These systems exhibit tunable secondary structures and result in sonication stimulus responsiveness of the organogels with the polypeptide segment variation, controlled by varying the ratio of BLG NCA to Gly NCA during the copolymerizations. Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) studies indicate the α-helical component decreases while the β-sheet content increases systematically with a higher mole fraction of Gly in the polypeptide segment. The supramolecular assembly of β-sheet nanofibrils, having a tunable width over the range of 10.4–14.5 nm with varied BLG to Gly ratio, are characterized by transmission electron microscopy (TEM). The further self-assembly of these nanostructures into 3-D gel networks within N,N-dimethylformamide (DMF) occurs at low critical gelation concentrations (CGC) (lowest ca. 0.6 wt%). Increased BLG to Gly ratios lead to an increase of the α-helical component in the secondary structures of the polypeptide segments, resulting in wider and more flexible nanofibrils. The presence of α-helical component in the polymers enhances the stability of the organogels against sonication, and instantaneous gel-to-gel transitions are observed as in situ reconstruction of networks occurs within the gelled materials after sonication. In marked contrast, the β-sheet-rich gel, prepared from mPEG-b-PGly, exhibits an instant gel-to-sol transition after sonication is applied. The CGC concentration and stiffness of this mPEG-b-P(BLG-co-Gly) organogel system can be tuned by simply varying the percentages of α-helix and β-sheet in the secondary structures through control of the BLG to Gly ratio during synthesis. The mechanical properties of these organogels are studied by dynamic mechanical analyses (DMA), having storage moduli of ca. 12.1 kPa at room temperature. The injectability and self-healing capabilities are demonstrated by direct observation of the macroscopic self-healing behavior experiment.
 Please wait while we load your content...
Please wait while we load your content...

