
Theoretical study of CO catalytic oxidation on free and defective graphene-supported Au–Pd bimetallic clusters
Abstract
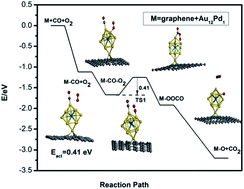
In this work, CO adsorption and oxidation on free and defective graphene-supported AumPdn (m + n = 13) clusters with either icosahedral (ICO) or truncated octahedral (TO) structures are investigated using density functional theory calculations. It is found that CO adsorbs more strongly than molecular O2 on the surface of these clusters, which is attributed to the strong hybridization between metal-d and C-p orbitals. In addition, the structural transformation from ICO to TO is observed for the Au-rich clusters upon CO and O2 adsorption because the stability of the ICO cluster is lower than that of the TO one. It is also found that the free Au12Pd1 cluster with TO structure exhibits the lowest energy barrier for CO oxidation among the free clusters (0.17 eV). When the Au12Pd1 cluster is supported on the single vacancy defective graphene, an increase in stability, a decrease in the adsorption strength of CO and O2, and a slight increase in the energy barrier (0.41 eV) for CO oxidation compared to the corresponding free cluster are observed. Our results are expected to be useful for future applications of graphene-supported bimetallic nanocatalysts.
 Please wait while we load your content...
Please wait while we load your content...

