
Utilizing thiol–ene coupling kinetics in the design of renewable thermoset resins based on d-limonene and polyfunctional thiols†
Abstract
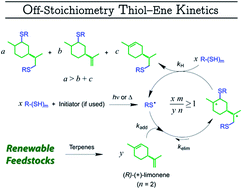
An extended model is developed to predict the free-radical thiol–ene reaction dynamics between D-limonene, as a renewable diolefin, and a monothiol compound (iso-tridecyl 3-mercaptopropionate) in bulk liquid conditions. Thermally and photo-initiated reactions of the two monomers showed favored thiol–ene coupling at the exo-isopropenyl alkene structure when reacted at 1 : 1 and 1 : 0.5 mole ratios. Experimental kinetic data obtained from the two stoichiometries were well reproduced numerically via the simulation software COPASI by introducing a multi-route mechanistic scheme with propagation–chain-transfer steps accounting for primary (mono-additions) and secondary (di-addition) modes of coupling. The differences in intrinsic double-bond reactivity enable synthesis of limonene-terminated resins (mono- versus poly-disperse) as multifunctional network precursors. Off-stoichiometry manipulations in the initial mole ratio, assisted by numerical simulations, offer a convenient approach to visualize the overall reaction system kinetics irrespective of temporal effects, thus being regarded as an important guiding tool for chemists aiming at designing thiol–ene systems based on limonene.
 Please wait while we load your content...
Please wait while we load your content...

