
Van der Waals density functional study of the energetics of alkali metal intercalation in graphite
Abstract
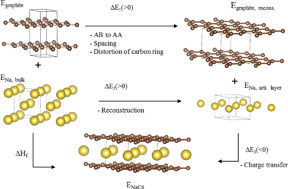
We report on the energetics of intercalation of lithium, sodium and potassium in graphite by density functional theory using recently developed van der Waals (vdW) density functionals. First stage intercalation compounds are well described by conventional functionals like GGA, but van der Waals functionals are crucial for higher stage intercalation compounds and graphite, where van der Waals interactions are important. The vdW-optPBE functional gave the best agreement with reported structure and energetics for graphite and LiC6 and was further applied for intercalation of Na and K. The enthalpy of formation of LiC6 and KC8 were found to be −16.4 and −27.5 kJ mol−1 respectively. NaC6 and NaC8 were unstable with positive enthalpies of formation (+20.8 and +19.9 kJ mol−1). The energetics of stacking of graphene and intercalant layers was investigated from first to fifth stage intercalation compounds. Higher stage compounds of Li and K were stable, but with less negative enthalpy of formation with increasing stage order. The higher stage Na compounds possessed positive enthalpy of formation, but lower in magnitude than the energy difference of 0.6 kJ mol−1 between graphite with AB and AA stacking. The abnormal behaviour of the lower stage Na intercalation compounds was rationalized by the lower energy involved in the formation of the chemical bond between carbon Na relative to the corresponding bond with Li or K. The chemical bond between alkali metal and carbon is characterized by charge transfer from the alkali-metal to carbon resulting in ionized alkali-metals. The intercalation induces only a subtle increase in the in-plane C–C bond lengths, with longer C–C bonds in the vicinity of the alkali metals but without breaking the hexagonal symmetry.
 Please wait while we load your content...
Please wait while we load your content...

