
Abstract
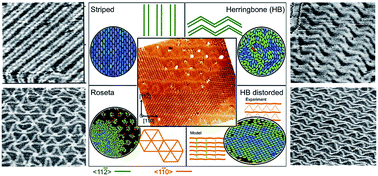
The prediction of supramolecular self-assembly onto solid surfaces is still challenging in many situations of interest for nanoscience. In particular, no previous simulation approach has been capable to simulate large self-assembly patterns of organic molecules over reconstructed surfaces (which have periodicities over large distances) due to the large number of surface atoms and adsorbing molecules involved. Using a novel simulation technique, we report here large scale simulations of the self-assembly patterns of an organic molecule (DIP) over different reconstructions of the Au(111) surface. We show that on particular reconstructions, the molecule–molecule interactions are enhanced in a way that long-range order is promoted. Also, the presence of a distortion in a reconstructed surface pattern not only induces the presence of long-range order but also is able to drive the organization of DIP into two coexisting homochiral domains, in quantitative agreement with STM experiments. On the other hand, only short range order is obtained in other reconstructions of the Au(111) surface. The simulation strategy opens interesting perspectives to tune the supramolecular structure by simulation design and surface engineering if choosing the right molecular building blocks and stabilising the chosen reconstruction pattern.
 Please wait while we load your content...
Please wait while we load your content...

