
An ab initio and DFT study of some homolytic substitution reactions of methoxycarbonyl radicals at silicon, germanium, and tin†
Abstract
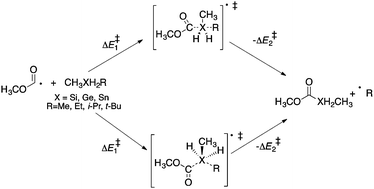
Homolytic substitution reactions of methoxycarbonyl radicals at the silicon, germanium, and tin atoms in various dialkylsilanes, dialkylgermanes and dialkylstannanes have been investigated using computational techniques. Ab initio and DFT calculations predict that attack of methoxycarbonyl radical at the silicon containing molecules can proceed via both backside and frontside attack mechanisms. At the (CCSD(T)/DZP//BHandHLYP/DZP) level of theory, energy barriers (ΔE‡) of 93.9 and 103.3 kJ mol−1 are calculated for the backside and frontside reactions, respectively. Similar results are obtained for reactions involving germanium and tin with calculated energy barriers (ΔE‡) of 89.2 and 99.2 kJ mol−1 for the backside and frontside attack (respectively) at germanium (CCSD(T)/DZP//BHandHLYP/DZP), and 71.8 (backside) and 71.9 kJ mol−1 (frontside) for the analogous reactions at tin. These data suggest that both homolytic substitution mechanisms are feasible for homolytic reactions of methoxycarbonyl radicals at silicon, germanium, and tin. These homolytic substitution reactions are also predicted to be endothermic at all levels of theory with the reverse reaction favoured by 11–37 kJ mol−1 for attack at silicon, 7–31 kJ mol−1 for attack at germanium, and by 9–30 kJ mol−1 for attack at tin, depending on the level of theory.
 Please wait while we load your content...
Please wait while we load your content...

