
Excitation wavelength dependence of the charge separation pathways in tetraporphyrin-naphthalene diimide pentads†
Abstract
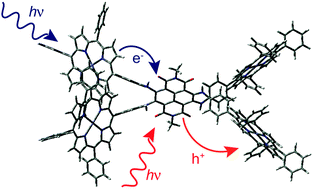
The excited-state dynamics of two multichromophoric arrays composed of a naphthalene diimide centre and four zinc or free-base porphyrins substituted on the naphthalene core via aniline bridges has been investigated using a combination of stationary and ultrafast spectroscopy. These pentads act as efficient antennae as they absorb over the whole visible region, with a band around 700 nm, associated with a transition to the S1 state delocalised over the whole arrays, and bands at higher energy due to transitions centred on the porphyrins. In non-polar solvents, population of these porphyrin states is followed by sub-picosecond internal conversion to the S1 state. The existence of a charge-separated state located above the S1 state could enhance this process. The decay of the S1 state is dominated by non-radiative deactivation on the 100 ps timescale, most probably favoured by the small S1–S0 energy gap and the very high density of vibrational states of these very large chromophores. In polar solvents, the charge-separated state lies just below the S1 state. It can be populated within a few picoseconds by a thermally activated hole transfer from the S1 state as well as via sub-picosecond non-equilibrium electron transfer from vibrationally hot porphyrin excited states. Because of the small energy gap between the charge-separated state and the ground state, charge recombination is almost barrierless and occurs within a few picoseconds. Despite their very different driving forces, charge separation and recombination occur on similar timescales. This is explained by the electronic coupling that differs considerably for both processes.
 Please wait while we load your content...
Please wait while we load your content...

