
Molecular dynamics simulations of ammonium/phosphonium-based protic ionic liquids: influence of alkyl to aryl group†
 *a
and
Anurag Prakash
Sunda
*b
*a
and
Anurag Prakash
Sunda
*b
Abstract
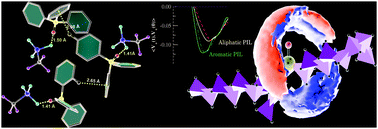
The variation of the center atom in the cation from an N to a P-atom leads to improved physiochemical properties of protic ionic liquids (PILs) which are suitable for electrolyte applications. We present an atomistic simulations study to compare the effect of an alkyl or aryl group on trioctylammonium triflate ([HN(Oct)3][TFO]) and triphenylammonium triflate ([HN(Ph)3][TFO]) with their phosphonium analogues. We have computed the binding energy from quantum chemical calculations and physical properties such as the viscosity and the electrical conductivity of PILs from molecular dynamics simulations. The influence of the aromatic character in PILs is found to be significant to the physical properties. Gas phase quantum chemical calculations on clusters of ion pairs have revealed the presence of C–H/π interactions in aromatic PILs along with hydrogen bonding. The variation in strength of the ion-pair affinities is examined using electric-current correlation and velocity autocorrelation functions. The qualitative differences observed are due to the aromatic rings and change in the central atom of the quaternary cation from an N to a P-atom, substantiated quantitatively by diffusion coefficients and electrical conductivities. The relatively weaker ion-pair interactions and low binding energy (−73.34 kcal mol−1) lead to the highest electrical conductivity in [HP(Ph)3][TFO].
 Please wait while we load your content...
Please wait while we load your content...

