
Hybrid density functional theory modeling of Ca, Zn, and Al ion batteries using the Chevrel phase Mo6S8 cathode
 a
and
Manuel
Smeu
*a
a
and
Manuel
Smeu
*a
Abstract
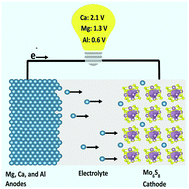
Hybrid density functional theory (DFT) is used to study the Chevrel phase Mo6X8 (X = S, Se, Te) as a promising cathode material intercalated with various metal ions (M = Li, Na, Be, Mg, Ca, Sr, Ba, Zn, Al). Electronic properties and voltages are calculated for each case. Ca ions are predicted to produce a voltage output ranging from 1.8–2.1 V, comparable to the voltage calculated for Li ions while providing two electrons per transferred ion. The highest voltage is determined to result when the chalcogen X in Mo6X8 is S, over Se or Te. Additionally, a comparison of the local-density approximation (LDA), the Perdew–Burke–Ernzerhof (PBE), the Hubbard U corrected GGA-PBE (PBE+U), the meta-GGA modified Becke–Johnson (mBJ), and the hybrid Heyd–Scuseria–Ernzerhof (HSE) functionals are made. The electronic structure determined with HSE is taken as the most reliable, and PBE and LDA can provide reasonable approximations. The PBE+U approach yields an erroneous band gap and should be avoided. The voltages calculated with HSE are in excellent agreement with available experimental data.
 Please wait while we load your content...
Please wait while we load your content...

