
The photochemistry of inverse dithienylethene switches understood†
Abstract
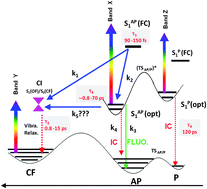
The photophysical properties of a series of dithienylethenes, free or blocked in an ideal photoactive conformation by an alkyl bridge, have been investigated by stationary, ultrafast spectroscopy and state-of-the-art time-dependent density functional theory calculations. Thanks to the clear ultrafast transient signatures corroborating NMR results, we bring strong evidence that the unreactive parallel open form conformer has been efficiently removed by the chain. For the first time, the photophysics of this species, namely an internal conversion of 120 ps is highlighted. In contradiction to the main ideas in the literature, the photocyclization mechanism is rationalized by a direct photocyclization mechanism from the Franck–Condon region passing directly through a conical intersection within ≈100 fs (not few picoseconds) while a competitive mechanism occurs through the relaxed S1 state. Relaxation processes (fluorescence and internal conversion) originating from this relaxed state are sensitive to the length of the blocking chain. Both concomitant pathways are necessary to rationalize: (i) the inverse relationship between emission and cyclization quantum yields and (ii) the non-unity value of the latter for bridged compounds.
 Please wait while we load your content...
Please wait while we load your content...

