
Vibrational spectra and structures of neutral Si6X clusters (X = Be, B, C, N, O)†
Abstract
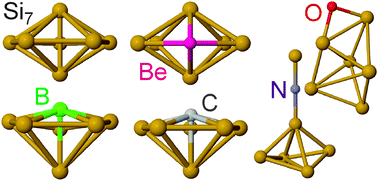
Neutral silicon clusters doped with first row elements (Si6X) have been generated (X = B, C, N, O) and characterized by infrared–ultraviolet (IR–UV) two-photon resonance-enhanced ionization spectroscopy (X = C, O) and quantum chemical calculations (X = Be, B, C, N, O, Si). In the near threshold UV photoionization, the ion signal of specific cluster sizes can be significantly enhanced by resonant excitation with tunable IR light prior to UV irradiation, allowing for the measurement of the IR spectra of Si7, Si6C, and Si6O clusters. Structural assignments are achieved with the help of a global optimization procedure using density functional theory (DFT). The most stable calculated structures show the best agreement between predicted and measured spectra. The dopant atoms in the Si6X clusters have a negative net charge and the Si atoms act as electron donors within the clusters. Moreover, the overall structures of the Si6X clusters depend strongly on the nature of the dopant atom, i.e., its size and valency. While in some of the Si6X clusters one Si atom in Si7 is simply substituted by the dopant atom (X = Be, B, C), other cases exhibit a completely different geometry (X = N, O). As a general trend, doping of the Si7 cluster with first-row dopants is predicted to shift the optically allowed electronic transitions into the visible or even near-IR spectral range due to symmetry reduction or the radical character of the doped cluster.
 Please wait while we load your content...
Please wait while we load your content...

