
Evaluation of composite schemes for CCSDT(Q) calculations of interaction energies of noncovalent complexes†
Abstract
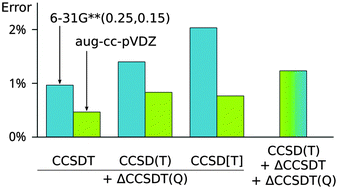
Recently, it has become possible to apply higher-order coupled-cluster methods to polyatomic systems including molecular noncovalent complexes. Due to the steep scaling of the complexity of these calculations, the size of the basis set becomes a critical factor and larger systems can be calculated only in small basis sets. To obtain the most accurate results, it is necessary to use composite schemes where the higher-order terms are added to a baseline calculation for which a larger basis set can be used. In this work, we have examined the accuracy of composite schemes where CCSDT(Q) correction calculated in a smaller basis set is added to CCSD(T), CCSD[T] and CCSDT calculations. As a benchmark, we have used CCSDT(Q)/aug-cc-pVTZ interaction energies calculated in a set of 18 small noncovalent complexes. We have found that the differences between the studied schemes are small and that it is safe to make the correction in a single step starting from the CCSD(T) baseline. The basis set dependence of the correction is strongly affected by the nature of the interaction. For dispersion-bound complexes, the correction calculated in a basis set as small as 6-31G**(0.25,0.15) improves the results consistently. On the other hand, description of polar complexes and especially hydrogen bonds is more difficult and the CCSDT(Q) correction has an incorrect sign until a rather large basis set is used; even the aug-cc-pVDZ result is not reliable in rare cases.
 Please wait while we load your content...
Please wait while we load your content...

