
The nature of bonding and electronic properties of graphene and benzene with iridium adatoms†
Abstract
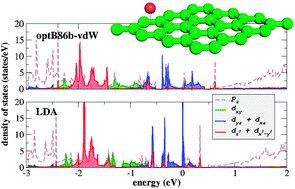
Recent theoretical simulations predicted that graphene decorated with Ir adatoms could realize a two-dimensional topological insulator with a substantial band gap. Our understanding of how the electronic properties of graphene change in the presence of metal adatoms is however still limited, as the binding is quite complex involving static and dynamic correlation effects together with relativistic contributions, which makes the theoretical description of such systems quite challenging. We applied the quantum chemical complete active space second order perturbation theory (CASPT2) method and density functional theory beyond the standard local density functional approach including relativistic spin–orbit coupling (SOC) effects on Ir–benzene and Ir–graphene complexes. The CASPT2-SOC method revealed a strong binding affinity of Ir for benzene (33.1 kcal mol−1) at a 1.81 Å distance, which was of a single reference character, and a weaker binding affinity (6.3 kcal mol−1) at 3.00 Å of a multireference character. In the Ir–graphene complex, the quartet ground-state of the Ir atom changed to the doublet state upon adsorption, and the binding energy predicted by optB86b-vdW-SOC functional remained high (33.8 kcal mol−1). In all cases the dynamic correlation effects significantly contributed to the binding. The density of states calculated with the hybrid functional HSE06 showed that the gap of 0.3 eV was induced in graphene by the adsorbed Ir atom even in scalar relativistic calculation, in contrast to metallic behaviour predicted by local density approximation. The results suggest that the strong correlation effects contribute to the opening of the band gap in graphene covered with the Ir adatoms. The value of the magnetic anisotropy energy of 0.1 kcal mol−1 predicted by HSE06 is lower than those calculated using local functionals.
 Please wait while we load your content...
Please wait while we load your content...

