
Correction of erroneously packed protein's side chains in the NMR structure based on ab initio chemical shift calculations†
Abstract
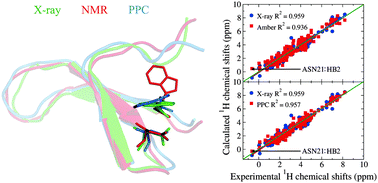
In this work, protein side chain 1H chemical shifts are used as probes to detect and correct side-chain packing errors in protein's NMR structures through structural refinement. By applying the automated fragmentation quantum mechanics/molecular mechanics (AF-QM/MM) method for ab initio calculation of chemical shifts, incorrect side chain packing was detected in the NMR structures of the Pin1 WW domain. The NMR structure is then refined by using molecular dynamics simulation and the polarized protein-specific charge (PPC) model. The computationally refined structure of the Pin1 WW domain is in excellent agreement with the corresponding X-ray structure. In particular, the use of the PPC model yields a more accurate structure than that using the standard (nonpolarizable) force field. For comparison, some of the widely used empirical models for chemical shift calculations are unable to correctly describe the relationship between the particular proton chemical shift and protein structures. The AF-QM/MM method can be used as a powerful tool for protein NMR structure validation and structural flaw detection.
 Please wait while we load your content...
Please wait while we load your content...

