
Site-selective adsorption of phthalocyanine on h-BN/Rh(111) nanomesh†
Abstract
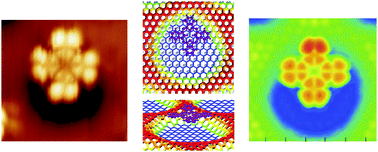
Experiment and computer simulations were conducted in order to study the adsorption of the phthalocyanine molecules H2Pc and CuPc on the h-BN/Rh(111) nanomesh. We combine STM investigations with the exploration of the potential energy surface as resulting from density functional theory calculations. Both approaches indicate a pronounced adsorption selectivity in the so called pore regions of the h-BN nanomesh, whereas the adsorption energy landscape in the pore turns out to be very shallow. This is seen by the inability to image the molecule stably at 77 K by scanning tunneling microscopy. Understanding the nature of the binding by rationalizing the site-selectivity and the mobility of the molecules is quite a challenge for both experiment and theory. In particular, we observe that the choice of the functional in the DFT description is crucial to be able to discriminate among adsorption sites that are very close in energy and to resolve low energy barriers. Our study reveals how the shape of the corrugated h-BN layer is the dominant factor that determines the subtle features of the potential energy surface for the adsorption of phthalocyanine.
 Please wait while we load your content...
Please wait while we load your content...

