
Halogen bonding of electrophilic bromocarbons with pseudohalide anions†
Abstract
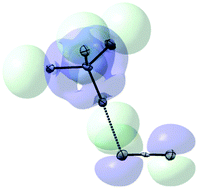
UV-Vis measurements showed that the interaction of pseudohalide anions, A− (A− = N3−, NCO−, NCS−), with electrophilic bromocarbons, R–Br (R–Br = CBr4, CBr3NO2, CBr3CONH2, CBr3H, CBr3F, CBr3CN or C3Br2F6), in solution results in formation of [R–Br, A−] complexes. These associates are characterized by intense absorption bands in the 200–350 nm range showing distinct Mulliken correlation with the frontier (HOMO–LUMO) orbitals’ separations of the interacting anion and the R–Br electrophile. X-ray crystallographic studies established the principal structural features of the halogen-bonded associates between bromocarbons and polydentate pseudohalide anions. Specifically, in the (Pr4N)NCO·CBr4, (Pr4N)N3·CBr4 and (Pr4N)NCO·CBr3NO2 co-crystals, bromine substituents of the electrophiles are halogen-bonded with the (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N or N
N or N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) π-bonds of the cyanate or azide anions. Co-crystals of CBr4 with (Pr4N)NCS show two modes (C–Br⋯S–C and C–Br⋯NC) of halogen bonding, while tribromoacetamide molecules form C–Br⋯S–C halogen bonds and N–H⋯NC hydrogen bonds with thiocyanate anions. Structures and energetics of the halogen-bonded complexes resulted from the M06-2X/6-311+G(dp) computations of various R–Br–A− pairs were consistent with the experimental data. These computations revealed that the variations of the intramolecular (C–Br) and intermolecular (Br⋯A−) bond lengths are correlated with the A− → R–Br charge transfer determined from Natural Bond Orbital analysis. Also, the scrutiny of the structural data indicated that the locations of the intermolecular contacts in these associates are determined primarily by the frontier orbital shapes of the halogen-bonded species. Thus, spectral and structural data point out a significant role of molecular-orbital (charge-transfer) interactions in formation of halogen bonded complexes involving pseudohalides and bromocarbons.
N) π-bonds of the cyanate or azide anions. Co-crystals of CBr4 with (Pr4N)NCS show two modes (C–Br⋯S–C and C–Br⋯NC) of halogen bonding, while tribromoacetamide molecules form C–Br⋯S–C halogen bonds and N–H⋯NC hydrogen bonds with thiocyanate anions. Structures and energetics of the halogen-bonded complexes resulted from the M06-2X/6-311+G(dp) computations of various R–Br–A− pairs were consistent with the experimental data. These computations revealed that the variations of the intramolecular (C–Br) and intermolecular (Br⋯A−) bond lengths are correlated with the A− → R–Br charge transfer determined from Natural Bond Orbital analysis. Also, the scrutiny of the structural data indicated that the locations of the intermolecular contacts in these associates are determined primarily by the frontier orbital shapes of the halogen-bonded species. Thus, spectral and structural data point out a significant role of molecular-orbital (charge-transfer) interactions in formation of halogen bonded complexes involving pseudohalides and bromocarbons.
 Please wait while we load your content...
Please wait while we load your content...

