
Principal component analysis of Mn(salen) catalysts†
Abstract
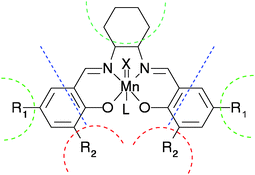
The theoretical study of Mn(salen) catalysts has been traditionally performed under the assumption that Mn(acacen′) (acacen′ = 3,3′-(ethane-1,2-diylbis(azanylylidene))bis(prop-1-en-olate)) is an appropriate surrogate for the larger Mn(salen) complexes. In this work, the geometry and the electronic structure of several Mn(salen) and Mn(acacen′) model complexes were studied using Density Functional Theory (DFT) at diverse levels of approximation, with the aim of understanding the effects of truncation, metal oxidation, axial coordination, substitution on the aromatic rings of the salen ligand and chirality of the diimine bridge, as well as the choice of the density functional and basis set. To achieve this goal, geometric and structural data, obtained from these calculations, were subjected to Principal Component Analysis (PCA) and PCA with orthogonal rotation of the components (rPCA). The results show the choice of basis set to be of paramount importance, accounting for up to 30% of the variance in the data, while the differences between salen and acacen′ complexes account for about 9% of the variance in the data, and are mostly related to the conformation of the salen/acacen′ ligand around the metal centre. Variations in the spin state and oxidation state of the metal centre also account for large fractions of the total variance (up to 10% and 9%, respectively). Other effects, such as the nature of the diimine bridge or the presence of an alkyl substituent in the 3,3 and 5,5 positions of the aldehyde moiety, were found to be less important in terms of explaining the variance within the data set. A matrix of discriminants was compiled using the loadings of the principal and rotated components that best performed in the classification of the entries in the data. The scores obtained from its application to the data set were used as independent variables for devising linear models of different properties, with satisfactory prediction capabilities.
 Please wait while we load your content...
Please wait while we load your content...

