
A hybrid MD-kMC algorithm for folding proteins in explicit solvent†
Abstract
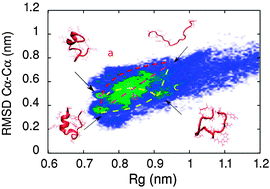
We present a novel hybrid MD-kMC algorithm that is capable of efficiently folding proteins in explicit solvent. We apply this algorithm to the folding of a small protein, Trp-Cage. Different kMC move sets that capture different possible rate limiting steps are implemented. The first uses secondary structure formation as a relevant rate event (a combination of dihedral rotations and hydrogen-bonding formation and breakage). The second uses tertiary structure formation events through formation of contacts via translational moves. Both methods fold the protein, but via different mechanisms and with different folding kinetics. The first method leads to folding via a structured helical state, with kinetics fit by a single exponential. The second method leads to folding via a collapsed loop, with kinetics poorly fit by single or double exponentials. In both cases, folding times are faster than experimentally reported values, The secondary and tertiary move sets are integrated in a third MD-kMC implementation, which now leads to folding of the protein via both pathways, with single and double-exponential fits to the rates, and to folding rates in good agreement with experimental values. The competition between secondary and tertiary structure leads to a longer search for the helix-rich intermediate in the case of the first pathway, and to the emergence of a kinetically trapped long-lived molten-globule collapsed state in the case of the second pathway. The algorithm presented not only captures experimentally observed folding intermediates and kinetics, but yields insights into the relative roles of local and global interactions in determining folding mechanisms and rates.
- This article is part of the themed collection: The free energy landscape: from folding to cellular function
 Please wait while we load your content...
Please wait while we load your content...

